![]()
Which method should I use to choose the alignment reference?
TransOmics™ Informatics provides three methods for choosing the alignment reference run, as seen below:
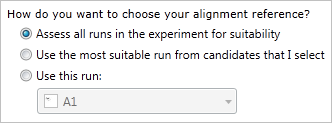
These three options and their use cases are described below.
1. Assess all runs in the experiment for suitability
This method compares every run in your experiment to every other run for similarity.
The run with the greatest similarity to all other runs is chosen as the alignment reference.
When should I use this method?
If you have no prior knowledge about which of your runs would make a good reference, then this choice will normally produce a good alignment reference for you.
This method can take a long time, however, as the running time is proportional to the square of the number of runs in your experiment. Below are some benchmarks for comparison:
| Number of runs | Time for reference selection |
|---|---|
| 2 | 0m 11s |
| 4 | 1m 2s |
| 6 | 2m 42s |
| 8 | 6m 54s |
| 10 | 10m 56s |
| 12 | 16m 38s |
Reference selection benchmarks, ran on a 6 core, 3.3 GHz, 16GB machine using profiled Thermo .raw files of average size 650MB.
If you have a large number of runs, therefore, you may wish to save time by choosing a subset of your runs as reference candidates.
2. Use the most suitable run from candidates that I select
This method asks you to choose a selection of reference candidates, and the automatic algorithm chooses the best reference from these runs.
When should I use this method?
This method is a good fit when you have some prior knowledge of the suitability of your runs as references, and you have multiple potential references in mind.
A couple of examples are:
- You have multiple runs from pooled samples.
- You know from knowledge of your experiment that runs from one experimental condition will contain the largest set of common peptides.
3. Use this run
This method allows you to manually choose the reference run.
When should I use this method?
This method is suitable when you want to choose the reference run yourself, for example, if you have a single run from a pooled sample that you wish to use.
Manual selection gives you full control, but there are a couple of risks to note:
- If you choose a pending run which subsequently fails to load, alignment will not be performed.
- If you choose a run before it fully loads, and it turns out to have chromatography issues, alignment will be negatively affected (for this reason we recommened that you let your reference run fully load and assess its chromatography before loading further runs).
Summary of methods
The table below gives a summary of the different methods:
| Method | When to use | Pros | Cons |
|---|---|---|---|
| Assess all runs in the experiment for suitability | No knowledge of which runs would make good references | Will normally choose a good reference without manual intervention | Can take a long time (especially if you have a lot of runs; Running time ∝ square of number of runs). |
| Use the most suitable run from candidates that I select | Some knowledge of which runs would make good references (e.g. runs from pooled samples, runs from a certain experimental condition) | Faster than considering all runs; algorithm will prefer runs with good chromatography | Potential to miss a better reference if your assumptions are incorrect; slower than manual selection (Running time ∝ number of runs × number of candidates). |
| Use this run | You know which run you wish to use as the reference (e.g. a single run from a pooled sample) | Fastest method and gives you full control | Alignment will be skipped if the run fails to import; bad chromatography will reduce alignment quality |