




How does normalisation work in Progenesis LC-MS?
The goal of normalisation is to determine a global scaling factor. There are several ways to achieve this and almost all normalisation methods used for proteomics and genomics data can be reduced to solving this problem. For example, spiking in known quantities of proteins, total protein abundance normalisation, variance stabilisation normalisation (VSN; also calculates an additive constant), ratiometric normalisation (with an internal standard). All techniques will need to deal with technical variation that can bias the calculation of the scaling factor. This can be modelled as follows:
y'i = αkyi
If you look through the details of these algorithms it quickly becomes apparent that the approach we follow is more robust and less influenced by noise in the data.
To implement normalisation we first select one run as a reference. Then for each feature we calculate a quantitative abundance ratio between the run being normalised and the reference run. The basic idea is that many proteins (and therefore peptide ions) won't be changing in the experiment and so the quantitative abundance ratio should equal to 1. Due to experimental (or technical) variations such as differences the quantity of protein loaded into the instrument, differences in ionization etc the actual ratio will not be 1. You need to correct for these experimental variations. This can be done by calculating a robust distribution of all ratios and determine a global scaling factor. The distribution is calculated using log(ratio) values and as log(1) = 0.0 we expect to see the ratio distribution scattered about 0.0. This ensures that up-regulation and down-regulation has the same weight. For example, given a ratio of 2/3 and a ratio of 3/2, you find that log(3/2) = +0.176 and log(2/3) = -0.176, so both are the same distance from 0 in log space. This is not the case in real space as 2/3 is closer to 1 than 3/2. We use the median and also use the median absolute deviation as an approximation of the variance and iterate this process to remove the influence of outliers. This gives a more robust estimation than just using the mean. The scaling factor is the anti-log of the average of the log(ratios).
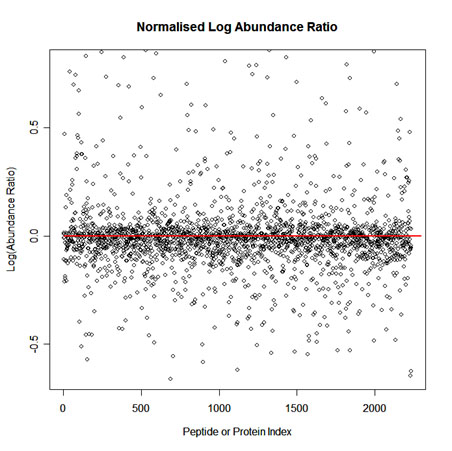
A scatter plot of Log(Abundance Ratio) after normalisation
In effect, you can think of each non-changing peptide ion as a "spike" and this will result in potentially thousands of "spikes" across the dataset to draw strength from when determining the scaling factor. Instead of a handful of these spikes, we use all the experimental data, again, a more robust approach.
A final point is that this approach to normalisation would be difficult if not impossible without prior high quality alignment and the detection of a single peptide ion detection map that is then applied to all runs in the sample, resulting in no missing values and 100% matching of data across all runs.