




Why does Progenesis LC-MS quantify before identifying?
The "quantify and then identify" approach promotes identification of low abundance peptides, often the ones you're most interested in, which have a low probability of being selected for MS/MS scans in the data driven acquisition approach. This is illustrated in the diagram below.
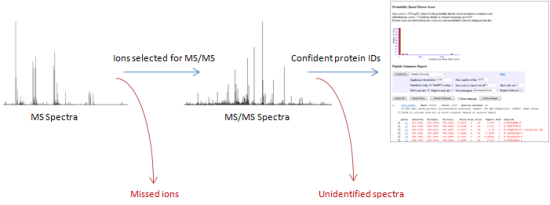
Information is discarded where data-dependent acquisition does not probe every ion. Also, any MS/MS spectra that are not confidently identified will be lost. This can result in significant protein behaviour being overlooked.
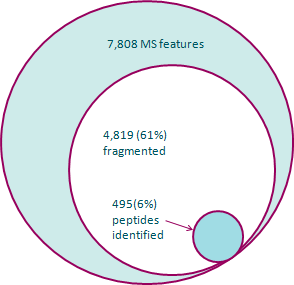
As an illustration of how much information can be discarded, the diagram at the left shows a breakdown of the potential data loss in a typical experiment. The outer ring represents the full set of features detected from the MS1 data. Inside that, it shows the number of those features that have MS/MS data recorded for them. Finally, the inner ring shows the features with a reliable identification. During the course of this, 94% of the original features have been discarded.
(Click on the image to view the exact numbers1.)
In Progenesis LC-MS, data is analysed at the LC-MS level to locate peaks exhibiting significant expression change between experiment groups.

By detecting and quantifying peptides at the MS level we don't miss any peptides that haven't triggered MS/MS scans. You can then export inclusion lists and re-run the sample immediately using the same established chromatography to generate MS/MS scans for any peptides missing this data. Because we quantify using the MS data the inclusion list can be focused on those peptide ions showing interesting behaviour. Also, you are still aware of the significant expression change and can investigate this further, even if a confident protein identification does not follow.
Two publications, The implications of proteolytic background for shotgun proteomics, Mol Cell Proteomics. 2007 Sep;6(9):1589-98. Epub 2007 May 28 and Comparative LC-MS: A landscape of peaks and valleys, Proteomics, vol 8, Issue 4, Pages 731 - 749.published online: 22 Feb 2008 describe some of the benefits of this approach compared to MS/MS data driven analysis.
Acknowledgements
- Many thanks to Dr Robert Parker and Prof Haroun Shah at the Health Protection Agency, London, UK for providing the sample data used in illustrating data loss.