




Step 2. Finding interesting peptides
To investigate the changes happening between samples, we need to define which samples represent which conditions and the subjects from which the samples were taken. This allows us to directly compare protein expression in different conditions, analysing the samples in those conditions as a whole.
The way that we do this depends on both the origin of our samples and how they were treated. This is because that information will determine the correct statistical treatment. The Experiment Design Setup screen offers 2 options:
- Between Subject: samples from any given subject appear in only one condition e.g. control subjects versus subjects with different drug treatments.
- Within Subject: here, samples have been taken from a given subject under different conditions e.g. the same subject has been sampled over a period of time, or after one or more treatments.
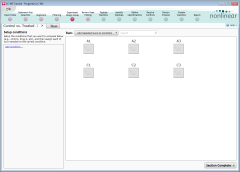 This tutorial experiment's design—control samples vs. treated—matches the first of these two, so:
This tutorial experiment's design—control samples vs. treated—matches the first of these two, so:
- Click anywhere within the Between Subject option.
- In the window that appears, enter Control vs. Treated in the Name field.
- Click the Create Design button.
We now need to group our control and treated samples:
- Click on each of the following runs to highlight them: A1, A2 and A3.
- Click the Add Selected Runs to Condition button. A new condition, named Condition 1, will appear in the list at the left.
- Click on the new condition's name to edit it; enter the name Control.
- Repeat the above 3 steps for the remaining runs, naming the second condition Treated.
Your experiment design should now look like this. Click Section Complete to continue.
{kind=link}
Finding statistically significant differences
 The Review Peak Picking screen is a powerful, and visually rich, screen that helps
you to:
The Review Peak Picking screen is a powerful, and visually rich, screen that helps
you to:
- Find features that show interesting variation between conditions
- Mark—or ‘tag’—and filter those features
- Review and, where needed, edit the results of peak picking
For simplicity, we'll skip the review and editing of the results of peak picking, and we'll concentrate on finding the interesting features. For this experiment, we want to identify features whose abundance is signficantly greater in the treated samples compared to the control samples.
We'll start by finding features whose abundance changes significantly either way:
- Right-click on any feature in the list at the left of the screen.
- From the pop-up menu that appears, select the Anova p-value… option in the Quick Tags sub-menu.
- When the New Quick Tag window appears, accept the default criteria of ≤ and 0.05, as well as the default name and tag color, and click Create tag.
All features that have an Anova (p) value of less than or equal to 0.05 will now have a red tag in the list's Tag column, as seen here:
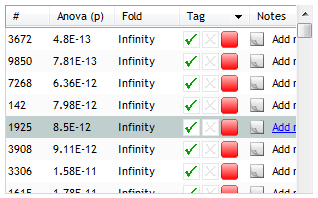
Next, we'll find features whose change in expression is greater than two-fold. While such a measure will depend greatly on your particular experiment, we'll use it here for the purposes of demonstrating the use and application of multiple tags.
- Once again, right-click on any feature in the list at the left of the screen.
- From the pop-up menu, select the Max fold change… option in the Quick Tags sub-menu.
- When the New Quick Tag window appears, accept the default criteria of ≥ and 2, as well as the default name and tag color, and click Create tag.
Tagging features, as we have done here, is the primary way of creating a list of peptides that require further attention. However, we also need to filter the list of features based on the tags we create:
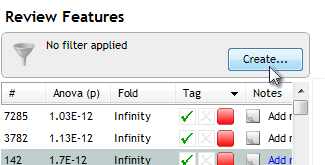
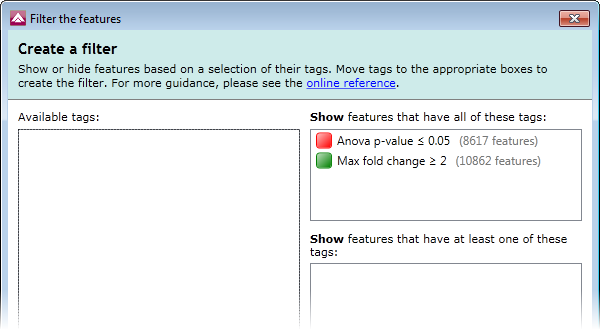
- Click the Create... button in the grey filter box above the list of features. This will display the tag filter window.
- In the Create a filter window, click on the Anova p-value ≤ 0.05 tag and drag it to the top box on the right-hand side: Show features that have all of these tags.
- Repeat the above step for the Max fold change ≥ 2 tag, dragging it into the top-right box alongside the first tag (show me).
{kind=link}
{kind=link}
This filter will result in the list of features showing only those that have both of these tags. Click OK to apply the filter. This updates the list accordingly and from this point onwards, we will work with only these significantly changing peptides.
Checking for consistent behaviour within conditions
Before we move on to the identification of our peptides, it's a good idea to use the Peptide Statistics screen to perform a quick bit of quality control. Specifically, we can use Principal Components Analysis (“PCA”) to check that, within a given experimental condition, the runs share the same overall behaviour.
- Click the Section Complete button to progress to the Peptide Statistics screen.
- As the screen loads, it will begin calculating a PCA plot of our filtered features. This should take only a few seconds at most.
Once complete, the upper half of the Peptide Statistics screen shows a PCA bi-plot. As well as showing the positions of the features on the principal component axes, the overall positions of the runs are shown. These are illustrated as small dots, using the same colour as the experimental condition from which they are taken. In our case, that's pink for Control samples and light blue for Treated samples.
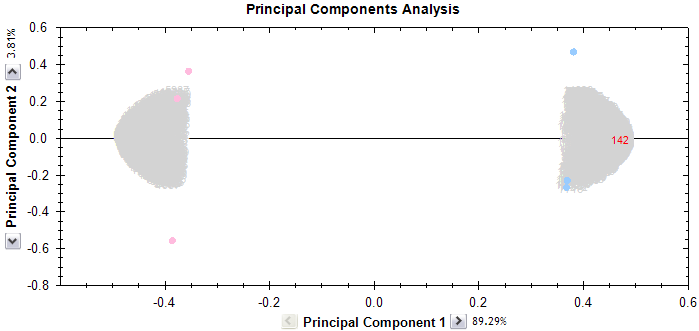
As we can see, the pink dots are clustered to the left and the blue dots are clustered to the right. If any of the dots had appeared as an outlier, this would have been a cause for concern. However, in our case, the clustering is as expected, so we can confidently continue our analysis.
Click the Section Complete button to move to the Identify Peptides screen, where we'll begin the process of identifying the peptides and proteins in our samples…