How can I use MSᴱ data to identify my peptides?
If you are using Waters MSᴱ data, you’ll be asked to configure the MSᴱ processing parameters after selecting files for import:
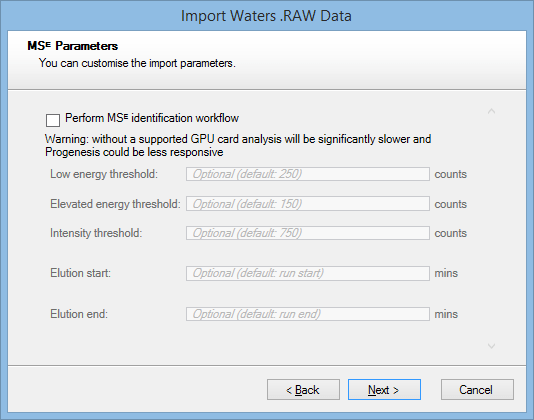
The MSᴱ identification workflow is selected by default for computers that have a supported GPU. On computers without such a GPU, the option is not initially selected, as software performance will be significantly slower.
When the MSᴱ workflow is selected, the default parameters will be used if no values are entered into the fields:
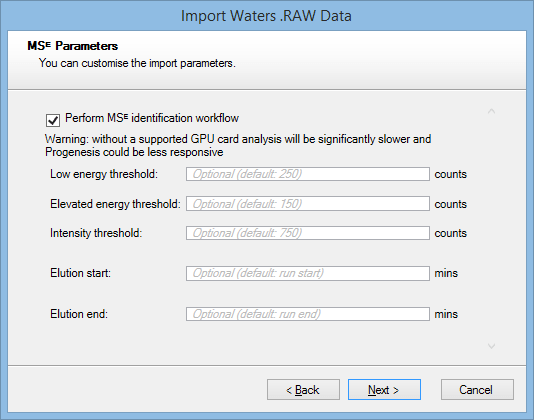
After making any necessary changes, click Next.
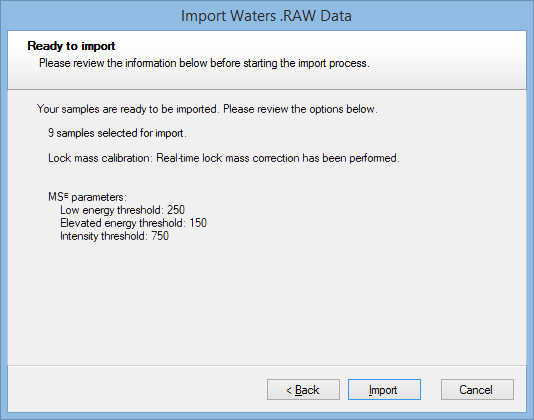
A window will then appear detailing the settings; if these are correct, select Import:
Once the file has imported, the necessary background processing required to perform the MSᴱ search may not be complete yet, but the software will allow you to continue in your analysis up until Identify Peptides. At this screen, after selecting the MSᴱ search peptide identification method, you will be able to determine whether your runs are ready for searching, as you should be able to see “x/n runs are ready for searching” in the pane to the left of the screen:
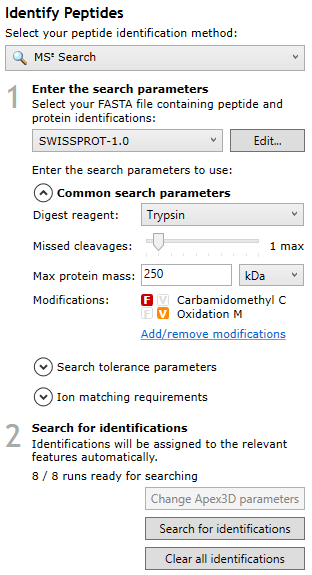
Note: for this background processing to complete, Progenesis must have access to the raw data — please do not remove the data from the location it was imported from until all runs are ready for searching.
Once all runs are ready for searching, you can select your search parameters using the options on the left of the screen — you can expand and collapse the options by selecting the arrow next to each set of parameters:
Common search parameters
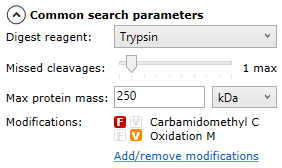
Search tolerance parameters

Ion matching requirements

Once you’ve made the required adjustments to the parameters, select Search for Identifications to start the search:
When the search is complete, a message will inform you of how many search hits have been retrieved: