




Exporting peptide measurements for external analysis
Within Progenesis QI for proteomics, deconvolution is used to group together peptide ions that are calculated to be of the same peptide. There are two exports that allow you to export this peptide (or, deconvoluted peptide ions) data. Similar to exporting peptide ion measurements, these two exports cover the pre-identification and post-identification stages within your workflow. The content of these two exports are referred to as deconvoluted peptide ion data and peptide measurements, respectively. As you'll read below, the former of these exports can include identification data.
Deconvoluted peptide ion data
This export allows external analysis at the peptide-level even if the ions have not been identified. It is available at the Review Peak Picking, Peptide Ion Statistics and Identify Peptides windows; access this export by selecting File | Export deconvoluted peptide ion data... as shown below:
This export provides similar data to that of the peptide ion data export, including the nature of the peptide itself (including the identifier of each peptide ion from the peptide), the inter-group differences associated with the peptide and its variability, and quantitative peptide data. The identifier used for a peptide is the combination of its retention time and neutral mass. For further details, see How are peptide values calculated from peptide ions? The export can be customised using the list shown below.
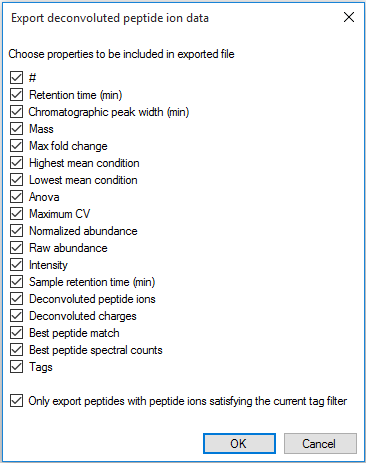
The column selection dialog box for Export deconvoluted peptide ion data.
Peptide measurements
In this peptide-level export each row is an identified peptide; only the peptide ions in your experiment that have identifications are deconvoluted to provide the data for this export. Deconvolution selects the best identification for each peptide.
Peptide measurements can be exported at the Review Proteins and Protein Statistics windows, via File | Export peptide measurements... as well as the Export peptide measurements button in step 4, at Review Proteins.

The peptide identifier is constructed in the same way as that of the deconvoluted peptide ion data export, being a combination of retention time and neutral mass. Similarly, the export includes data relating to the nature of the peptide itself (including the identifier of each peptide ion from the peptide), the inter-group differences associated with the peptide and its variability, and quantitative peptide data. For further details, see How are peptide values calculated from peptide ions? The export can be customised using the list shown below.
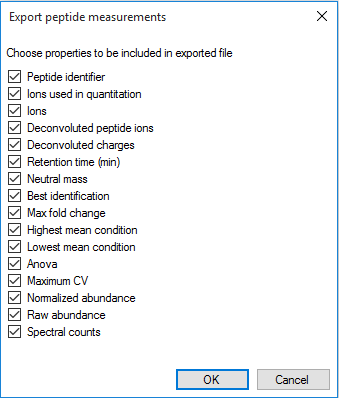
The column selection dialog box for Export peptide measurements.