




How can I detect more ions in my samples?
There are 2 sections of the workflow in Progenesis that control how many ions are detected in your data: the first is the filtering performed at Import Data; and the second is the peak picking parameters used at Peak Picking.
Making changes to the filtering strength at Import Data
We’ll start with how you can edit the filtering at Import Data. When data is imported into Progenesis, it is subjected to a filter which helps to strip out any background noise in your runs – on occasion you might want to adjust this setting:
- Start a new experiment in Progenesis QI for proteomics as you would normally, selecting the appropriate data capture settings.
- Before you import your files, at the Import Data screen, select Import data settings from the File menu to bring up the following window:
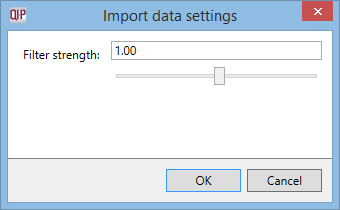
- Edit the new filter strength required – the setting of 1 is the default strength, a setting of 0 is no filtering applied, and a setting of 0.5 is half the strength of default. Once you’ve entered the new filtering strength, click OK.
- Import all of your runs as you would normally – the modified filtering strength will appear in the top right hand corner in the About this run section:

It’s important to note that this modified filter strength will only apply to data selected to be imported after the changes have been made, and only to the runs in this experiment. Also that all runs in an experiment should be filtered at the same strength – a warning will appear if you’ve used mixed filter strengths:

This article is intended for use where you wish to increase the number of ions detected in your data, but should you wish to reduce the number of detected ions for data sets with high levels of background noise, the same instructions apply. For example, a setting of 2 is equivalent to 2x default filter strength and will accordingly result in fewer ions detected.
Increasing the sensitivity of peak picking
The default settings used at peak picking aim to detect as many “true” ions as possible while ignoring any potential noise or contamination. On occasion, however, it may be necessary to edit these settings in order to detect more (or fewer) ions.
After your runs have been aligned successfully, you can move on to the Peak Picking stage of the workflow. If you wish to change the parameters used, click on Change parameters located on the left hand side of the screen to display the below window:
Click on the Peak picking limits tab:
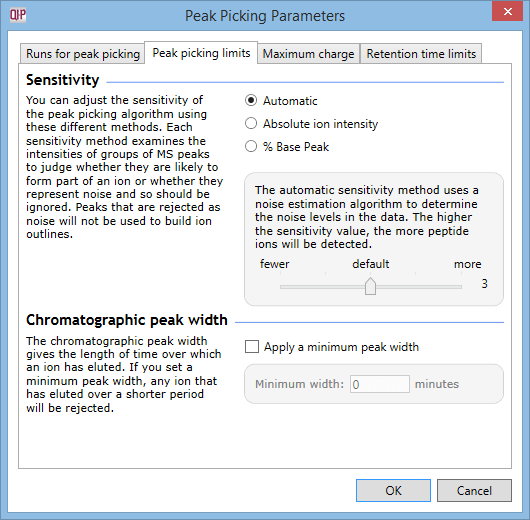
-
The top section relates to the sensitivity of peak picking with 3 options: Automatic, Absolute ion intensity, and % Base peak each with configurable settings.
The Automatic sensitivity method uses a noise estimation algorithm to determine the noise levels in the data. The higher the sensitivity level, the more peptide ions will be detected. The default setting is 3 and will go as high as 5 (and as low as 1).
For the Absolute ion intensity method, groups of peaks with an absolute intensity level less than the given value are ignored. Intensity levels from the aggregate run are used and the units, typically counts, are determined by your MS machine. Entering a value of 0 is the most sensitive setting for peak detection so will result in the maximum number of ions being detected.
Using the % Base peak option, peaks with an intensity less than a given percentage of the most intense peak are ignored. Intensity levels are compared in the aggregate run. Entering a value of 0 in here will also result in the maximum number of ions being detected.
-
Once you’ve chosen the appropriate method and made changes as necessary, click OK to close the window.
Note: If you’ve already carried out peak picking on this data set, you’ll be asked if you want to clear existing features and start again – click Clear peptide ions and start again.
To start the peak picking using your modified parameters, select Start peak picking.