




Answer your biological question with Progenesis QI for proteomics
Progenesis QI for proteomics is discovery analysis software for your LC-MS data; a revolutionary ‘difference engine’ that works in a unique way to help you to answer your biological question.
“We went from spending countless hours analyzing thousands of lines on spreadsheets, to completely automated quantitative proteomics performed in only a few hours. We also significantly improved our MS approach, as a result of the excellent QC insight offered by Progenesis.”
Paul Langlais
University of Arizona, Arizona, USA

Below are some key benefits that make Progenesis QI for proteomics unique:
Find the biologically interesting expression changes in your data
- Progenesis finds the real expression changes in your data
- Absolute quantitation, for any instrument, enables within-sample protein comparison
Get results from data with a large amount of biological variation
- The unique Progenesis co-detection workflow maximises the ability to find expression changes in your data
- Scalable to meet complex experimental design
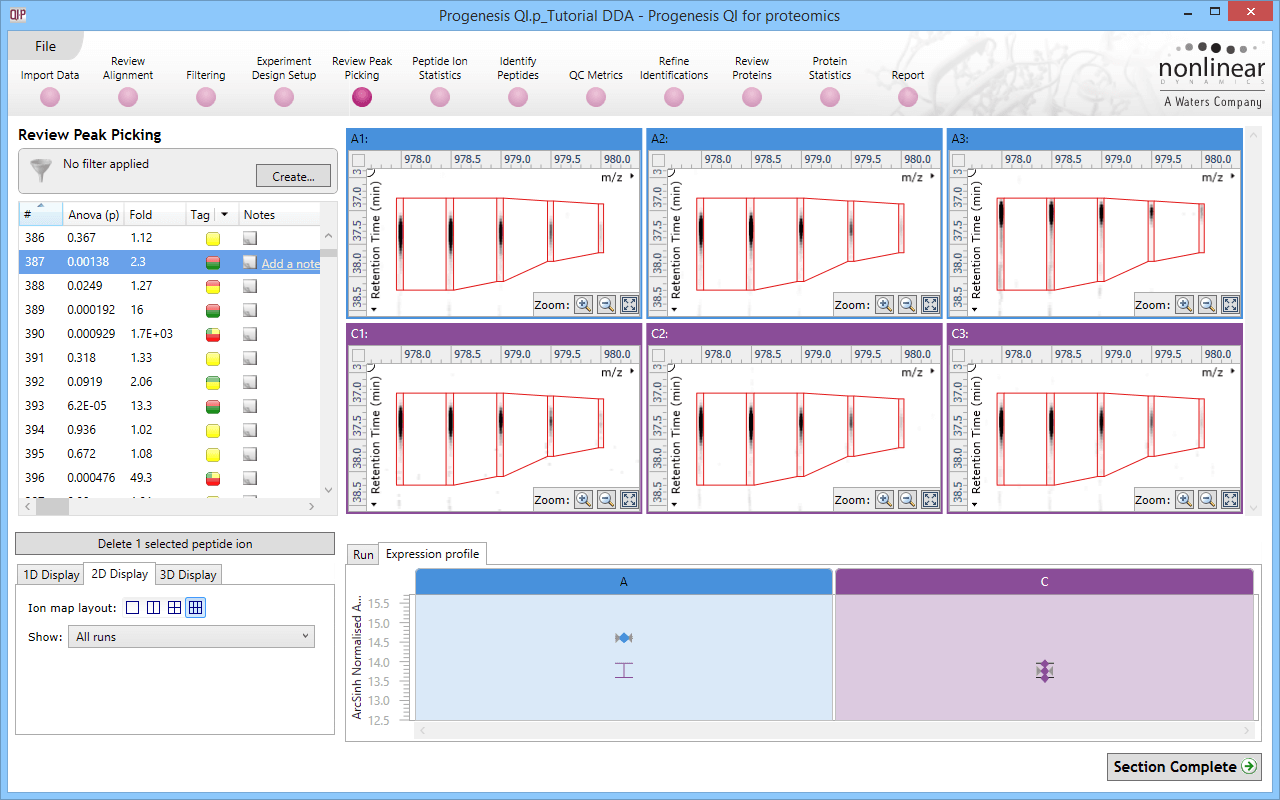 Click for full screenshot showing the results of co-detection
Click for full screenshot showing the results of co-detection
Multi-vendor, straightforward workflow with visualisation at every step
- Ease-of-use and visualisation make sense of very complex data at each analysis step
- Significantly reduces training time and can be used across all the mass spectrometers in an institute
Converts peptide-based data to protein expression data
- Put results into biological context using multivariate statistics and pathway analysis
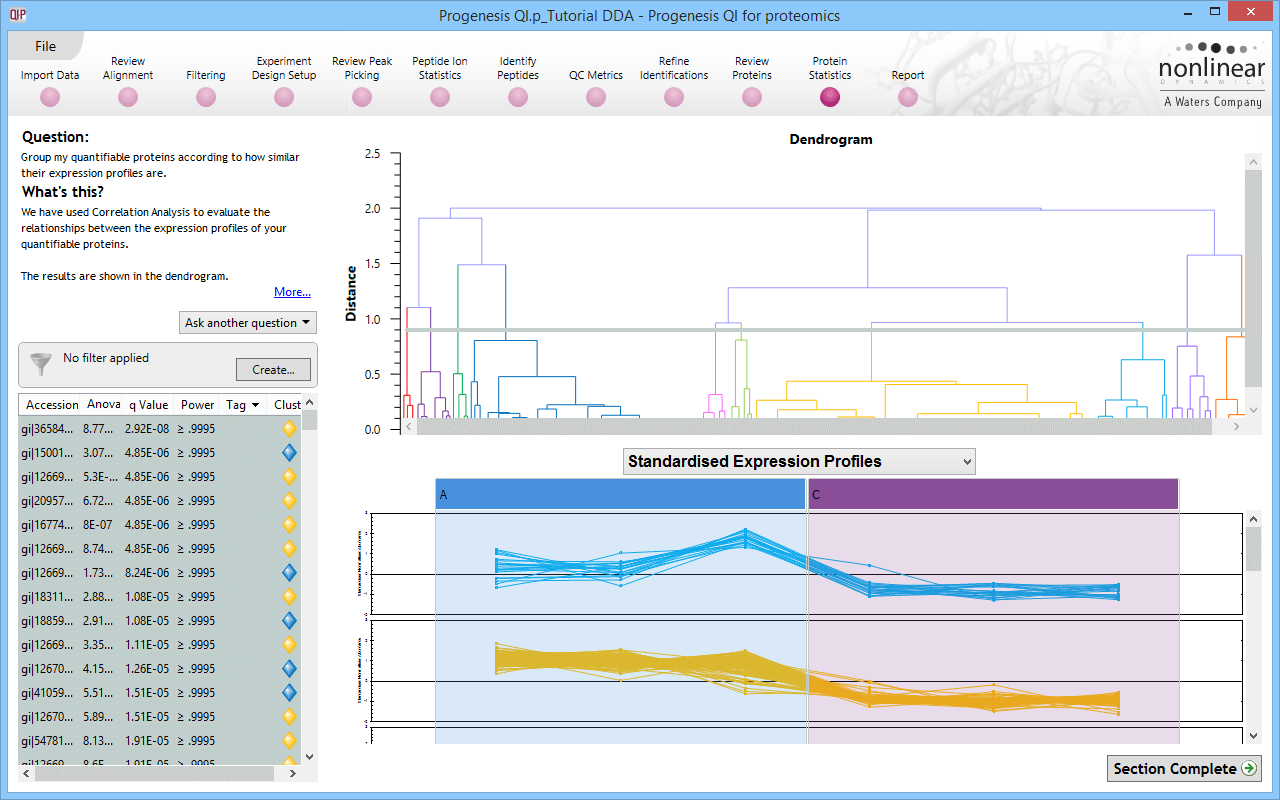 Click for full screenshot showing correlated protein expression profiles
Click for full screenshot showing correlated protein expression profiles
Quality control gives you confidence in your experimental conditions, instrument set up and data analysis
- The transparent Progenesis workflow, with its clear data visualisations, means you can:
- Assess the suitability of your data from any instrument at the very start of your analysis
- Understand your data and the analysis process
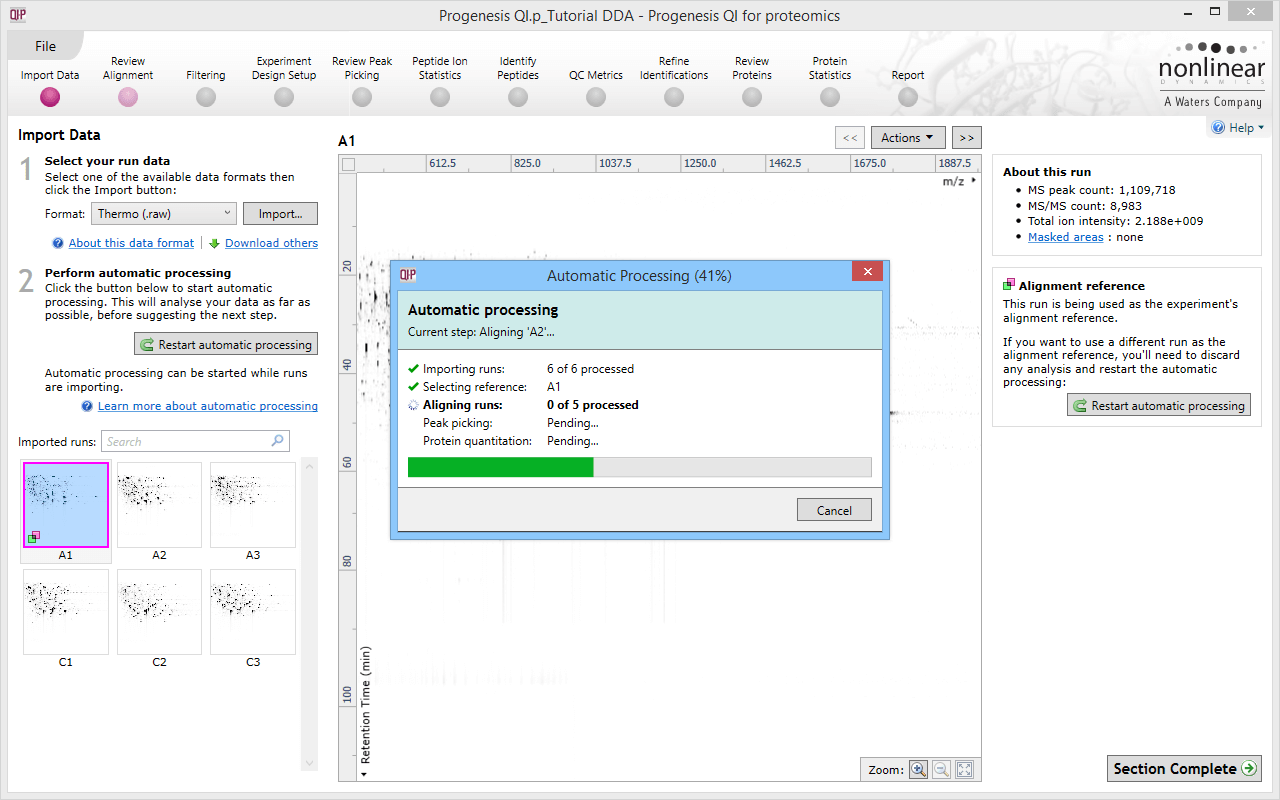 Click for full screenshot showing the start of the workflow in Progenesis
Click for full screenshot showing the start of the workflow in Progenesis
So, you have seen the benefits, now understand how it works. You can download Progenesis QI for proteomics and try it for yourself, on your own data.