




How can I get more identifications using inclusion lists in Progenesis?
An inclusion list can be used to target the collection of MS/MS data from additional LC/MS runs. This is a 2 step process: the first step is to export the inclusion list from Progenesis; the second step is to add the new runs to your existing analysis to associate their ms/ms spectra with the features already found by peak picking.
Step 1: Exporting the inclusion list
- At the Identify Peptides screen, filter your data to show only those ions for which you wish to collect additional fragmentation data. The quickest way to do is through the use of Quick Tags, specifically the No MS/MS data tag.
- To create an inclusion list for these tagged ions, go to File | Export inclusion list… to display the following window:
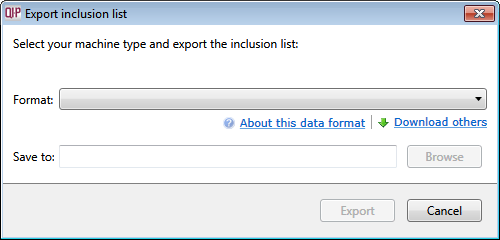
- Select the appropriate instrument type from the inclusion list and then choose where the file should be saved using the Browse button.
- Click Export
Note: For Thermo Finnigan inclusion lists, you’ll be asked if you want to widen the retention time windows:
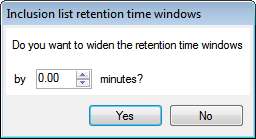
Make adjustments as necessary and then click either Yes or No, dependent on whether you wish to widen the retention time windows. - Use the inclusion list(s) to generate additional LC/MS runs, ensuring they are run under the same conditions as the original runs in the analysis.
Step 2: Using the additional runs generated from your inclusion list(s)
- Go back to the start of the experiment by selecting Import Data at the top of the screen and import all of the additional runs. After import, click Section Complete.
- Run Automatic Alignment for the additional run(s).
- Click Section Complete to move to the Filtering screen — the following window will display asking if you want to use the existing peak picking or replace the existing peak picking:
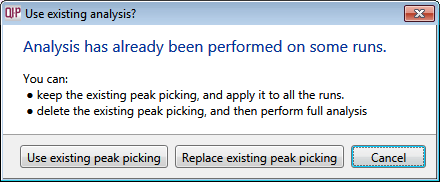
Select Use existing peak picking. - Once this has completed, you can skip forward to Identify Peptides where you can use the additional ms/ms spectra to aid your identifications — the features previously tagged as having No MS/MS data should now have some associated spectra, dependent on the success of your data collection.