How can I use MSᴱ, HDMSᴱ or SONAR data to identify my peptides?
If you are using Waters MSᴱ, HDMSᴱ or SONAR data, you can identify your peptides with the Ion Accounting identification workflow. You’ll be asked whether you want to use this workflow after selecting files for import:
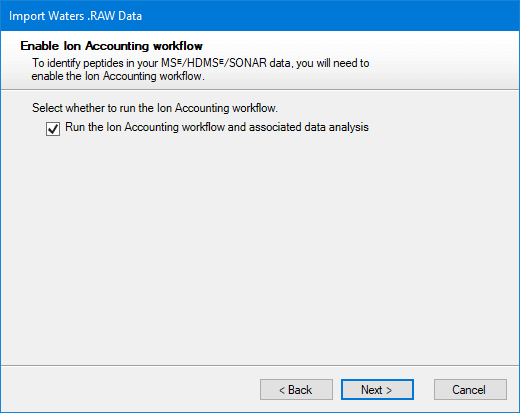
The Ion Accounting identification workflow is selected by default for computers that have a supported GPU. On computers without such a GPU, the option is not initially selected, as software performance will be significantly slower.
When the Ion Accounting identification workflow is selected, the next page will ask you how you want thresholds to be determined. This can be done automatically by Progenesis using the method described below. To use this feature you must specify a FASTA file. Alternatively, if you know what the right thresholds are for this run, you can specify them manually:
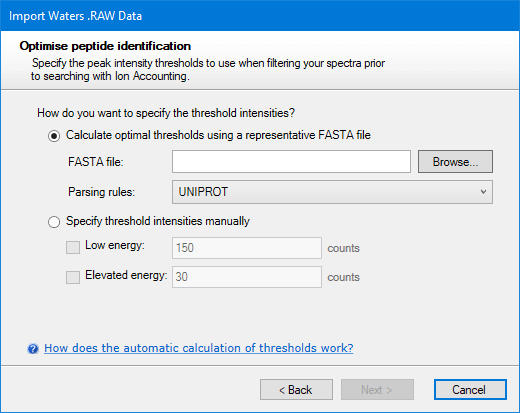
The manual option can be left with default values if you wish. After deciding which thresholds to use, click Next. You will then be asked to choose whether you want the Ion Accounting workflow applied to the whole run or only within a more limited range of elution times.
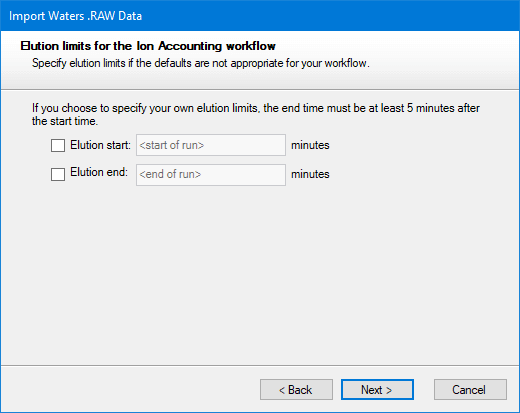
After making any necessary changes, click Next.
A window will then appear detailing the settings; if these are correct, select Import:
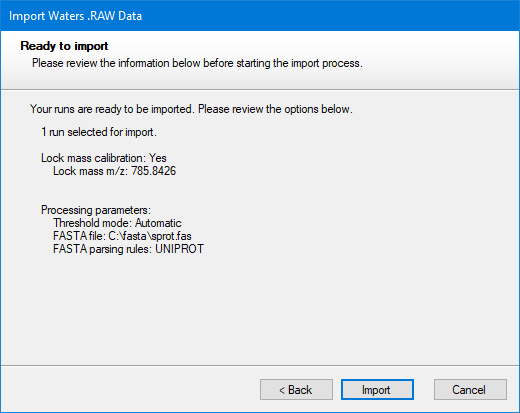
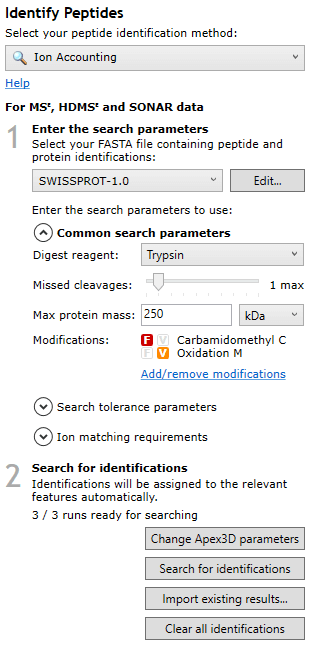
Once the file has imported, the necessary background processing required to perform the Ion Accounting search may not be complete yet, but the software will allow you to continue in your analysis up until Identify Peptides. At this screen, after selecting the Ion Accounting peptide identification method, you will be able to determine whether your runs are ready for searching, as you should be able to see “x/n runs are ready for searching” in the pane to the left of the screen:
Note: for this background processing to complete, Progenesis must have access to the raw data — please do not remove the data from the location it was imported from until all runs are ready for searching.
Once all runs are ready for searching, you can select your search parameters using the options on the left of the screen — you can expand and collapse the options by selecting the arrow next to each set of parameters:
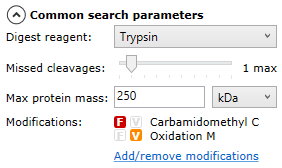
Common search parameters
Search tolerance parameters

Ion matching requirements

Once you’ve made the required adjustments to the parameters, select Search for Identifications to start the search:
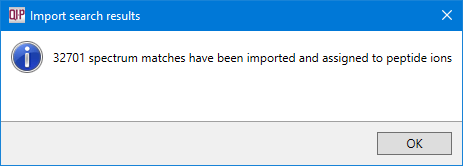
When the search is complete, a message will inform you of how many search hits have been retrieved:
How are thresholds calculated automatically?
Progenesis determines appropriate thresholds by sampling each run and finding the thresholds that yield the most protein identifications in the sample area. For each run, Progenesis performs the following steps:
- It finds the 5-minute retention time window that contains the highest total intensity.
- It extracts the ions within this window and performs multiple Ion Accounting searches, each one using a different set of threshold values.
- Finally, it selects the thresholds that resulted in the largest number of protein identifications and applies those to the whole run. If more than one set of thresholds results in similar numbers of identifications (within 10% of the maximum), it will choose the highest thresholds as a way of optimising system performance.
Importing existing results
If you have cleared the retrieved identifications and want to get them back without re-running the search, or otherwise have some Ion Accounting search result files
that you wish to import (e.g. having run the search externally), use the Import existing results button. You'll be prompted
to select a folder or folders containing the search result files. The inital folder shown will always be the current experiment's analysis folder, so if you
wish to quickly re-import the previous search results, you can simply accept the folder selection dialog there and then.
Progenesis will then look in the given folder or folders, and their subfolders, if any,
looking for files with names having the following form: <run-name>_IA_workflow.xml, and retrieve the search results from them, matching them
to the runs in the experiment based on the <run-name> part of the file name.
Searching with search engines other than Ion Accounting
It is possible, but not recommended, to search Waters MSᴱ, HDMSᴱ or SONAR data in search engines other than Ion Accounting. Where Ion Accounting is designed specifically to deal with these types of data, other search engines may not deal well with the inherent complexity.
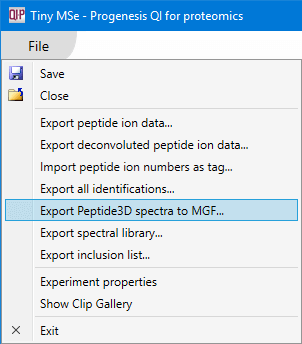
To obtain an MGF file searchable in other search engines, select the Export Peptide3D spectra to MGF... option from the File menu. If it hasn't already, you may want to wait for background processing to complete (the Search for identifications button is enabled), as only spectra from processed runs can be exported.
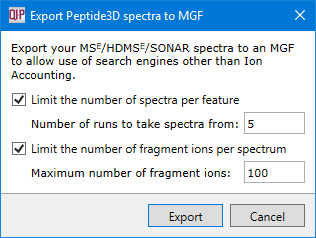
You will be able to select options designed to limit the amount of spectrum data exported.
- Limit the number of spectra per feature - Only export spectra from the configured number of runs in which a given feature is most intense.
- Limit the number of fragment ions per spectrum - Only export the configured number of most intense fragment ions in any given spectrum.
Together, they should help search engines not designed for MSᴱ, HDMSᴱ or SONAR data give better search results in a shorter amount of time.
After confirming the export options, click the Export button and select the location and name of the MGF file. The export will then proceed.
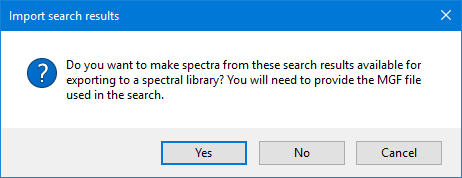
After the export completes, you can search the MGF file in an external search engine of your choice and import the results, provided it's supported by one of the peptide search plug-ins. If you wish to export these results and spectra to a spectral library, hold on to the MGF file, answer Yes when asked about it at the search results import stage, and select the MGF file used in the search. If you skip this step, the exported spectral library entries will contain no spectrum information, and the spectral library search engine will only be able to match them by mass.