




How are protein abundances calculated?
Detection and quantification of all peptide ions is followed by expression analysis and identification of the proteins from which they originate. Protein abundance is calculated from the sum of all unique normalised peptide ion abundances for a specific protein on each run. Alignment and co-detection of features means you have exactly the same number of quantified peptides identified on all runs so you can compare the sum of ion abundances between groups.
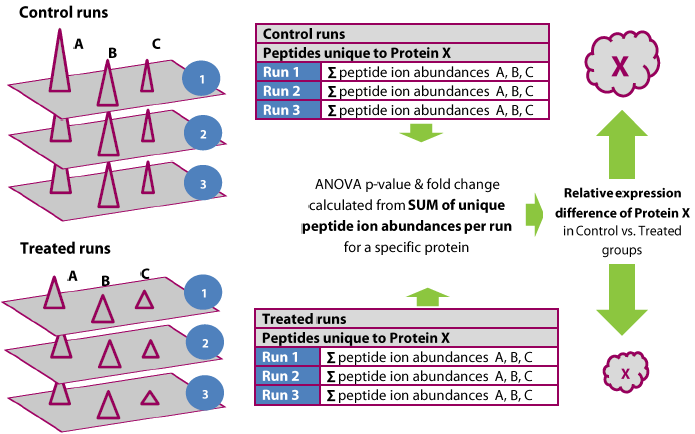
Features of protein quantification
- Co-detection of features provides ion abundance measures for the same peptides from each protein on every run, with no missing values
- Summing means that ion abundances with higher signal, and therefore less “noisy”, have more weight; making the final protein quantification more reliable
- When two protein identifications share common peptides the software uses unique peptides for quantification
- Searching peptides against databases can return multiple entries that are actually the same or related proteins, so the software automatically groups similar proteins into one quantification result
- Fractionated proteins are quantified in the same way but include all peptides from every fraction
For more information on the calculation of protein measurements, plus evidence of how it has been validated, please see the following technical note:
Protein Quantification by Progenesis LC‑MS.pdf (1MB)