




How are protein identifications grouped?
With bottom-up proteomics data, there is the challenge of handling redundancy in the matching of peptides to protein hits [1].
By the Parsimony Principle, protein hits may be reported as the minimum set that accounts for all observable peptides (the simplest explanation that fits all the facts is the most likely to be true, or no more assumptions should be made than necessary – a form of Occam’s razor). This may be applied to reduce the protein list where peptides could belong to several proteins.
For example, if Protein A is identified with five peptides, Protein B with two of those same peptides, and Protein C with the other three (see the first case in the table), it is simplest to assume that only Protein A is present using this logic – reporting only hits for which there is independent evidence and assuming one peptide corresponds to one protein source.
On the other hand, potential identities may be lost applying this approach. In the second case in the table, if protein D comprises peptides 1 and 2, protein E peptides 2 and 3, and Protein F peptides 3 and 4, then a strict parsimonious approach would only report proteins D and F, as the peptides for E can then be explained using only two entities as opposed to three.
| Peptides | |||||||
| Case 1 | 1 | 2 | 3 | 4 | 5 | Parsimony Reported? | Grouping Reported? |
|---|---|---|---|---|---|---|---|
| Protein A | X | X | X | X | X | Yes | Yes |
| Protein B | X | X | No | No | |||
| Protein C | X | X | X | No | No | ||
| Case 2 | 1 | 2 | 3 | 4 | 5 | Parsimony Reported? | Grouping Reported? |
| Protein D | X | X | Yes | Yes | |||
| Protein E | X | X | No | Yes | |||
| Protein F | X | X | Yes | Yes | |||
Examples of two protein sets and their handling under strict parsimonious and protein grouping approaches. See text for details.
Progenesis QI for proteomics does not apply a strict parsimonious approach as this may be over-stringent [2]. However, conflict resolution (as described in the user guide, page 42) may be carried out to refine the assignment of peptides to particular proteins directly if desired.
This links to the related principle of protein grouping, however, which Progenesis QI for proteomics does include. Where proteins are identified containing the same peptides then they are effectively indistinguishable aside from score, for example; the evidence is the same for both. Also, where one protein contains only peptides representing a subset of another protein’s peptides, that with fewer can be subsumed into that with the greater number. The first example in the table (A, B, C) would involve the subsuming of B and C into A under such a grouping approach. This is a less stringent approach, however, in that only subsets completely contained within another set would 'roll up'. In the second example in the table, Protein E would still be reported under a protein grouping approach as it is not wholly subsumable into either D or F.
This protein grouping approach can be enabled at the Protein options dialog at the Resolve Conflicts or Review Proteins windows:
The Protein options button in the Review Proteins window.
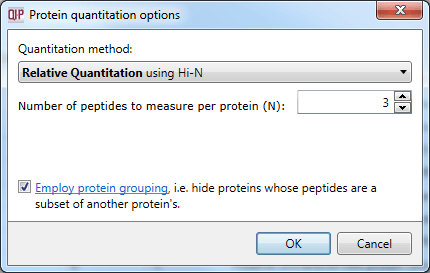
The protein grouping selection dialog within Protein options.
In practice, this causes protein identities derived from peptides that are a subset of an identification based on a larger set to be subsumed into that larger set. All relevant identities are listed as a group under the ‘lead’ identity with the greatest coverage (or highest score where coverage is equal). Quantitation is performed using the 'lead' identity peptide ion data only; potentially different results would arise from using the subsumed subsets, of course, but these are not calculated by definition under this approach.
The application of a protein grouping approach can simplify the data set, but again it may also be an oversimplification [2]; the user is able to make this choice as appropriate.
You can visualise the 'rolled-up' proteins by hovering over a protein accession at the Review Proteins window, for example. Where identities have been subsumed, the protein accession will have a symbol such as '+1' added (see below) and hovering over this will bring up details of the entire group.
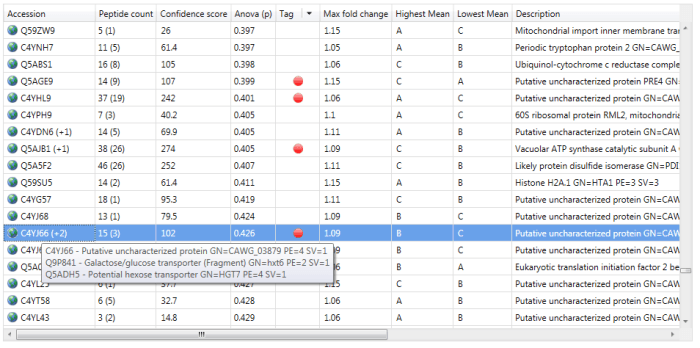
An example of a protein group. Quantitation is only based on the lead identity.
It is also possible to obtain the grouped accessions via exporting the protein measurements table as described here. In the export described at the linked FAQ, the subsumed identities are listed in the accession column in full.
Furthermore, double-clicking on a protein's row in the table or selecting the view peptide measurements link on the side bar will bring up the more detailed peptide measurements table for the protein. In this table, all the peptide ion (and peptide sequences) for that protein are shown. When grouping is active, naturally this will be the whole set for the identity with the greatest coverage. To see which peptide ions (and so peptides) belong to any 'rolled up' sub-identities, use the drop-down "Select ions of" menu that appears on the left when you examine a grouped protein entry in the table. The ions belonging to a selected sub-identity will be highlighted and may be tagged if desired.
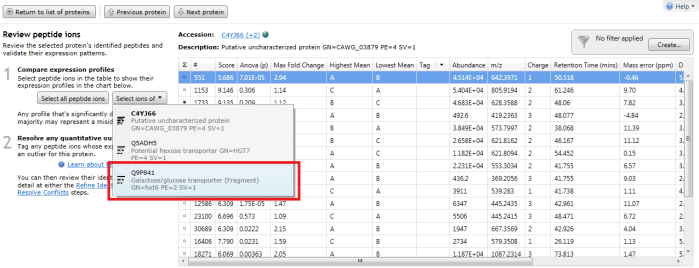
The peptide measurements table showing the "Select ions of" drop-down. In this case Q9P841 is the sub-identity being selected.
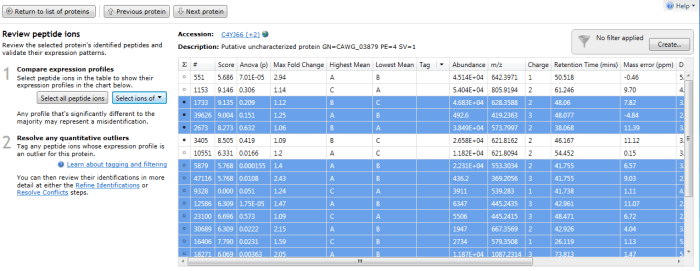
The peptide ions corresponding to Q9P841 are automatically highlighted.
Protein grouping and unique peptides
The protein grouping approach also has an important implication for quantifying proteins by unique peptides only. If proteins are not grouped, then peptide conflicts arise where the same peptide is used across multiple identities such as those that could be grouped, and this would eliminate them from use in quantitation under that scheme. Protein grouping removes this issue by counting the grouped peptides as unique (unless, of course, they overlap with another group's peptides).
References
- Nesvizhskii and Aebersold 2005: "Interpretation of shotgun proteomic data: the protein inference problem". Mol Cell Proteomics 4.10: 1419-40.
- Serang et al. 2012: "Recognizing uncertainty increases robustness and reproducibility of mass spectrometry-based protein inferences". J Proteome Res. 11 (12): 5586-91.