




How does absolute quantitation by Hi-N for HCP analysis work?
For Host Cell Protein (HCP) analysis, you can quantitate the absolute amounts of HCP contaminants in a run, by relating their measured abundance to that of a calibrant within the same run.
The basic process is similar to that for Absolute Quantitation using Hi-N, in that you nominate a calibrant protein for which you know the amount present in the sample, and the software will then use this information to calculate the absolute amount of other proteins that are identified in the analysis.
However, there are two differences:
- The mass of protein present (in ng) is now also provided for the calibrant and all other identified proteins, in addition to the amount in fmol.
- The calculation of protein amounts and masses present is now based on measurements of the calibrant protein present in the same run, and not a pooled measurement over all the runs. This is intended to make it simpler for you to relate amounts of HCPs to your therapeutic protein on a run-by-run basis.
In more detail on (2), the original method assumed that you had spiked a calibrant protein into your samples, and that this was present at the same amount and concentration in every sample. It then derived a relationship between protein amount and observed abundance using a pooled estimate from the calibrant across all the runs based on this assumption (more detail here). The HCP-specific option does still assume a specified amount of a calibrant in every run. However, it does not assume the same concentration and so does not pool information across the runs to derive its relationship between the calibrant and observed abundance. The amount of a contaminant is instead calculated directly by relation to the observed amount of the calibrant in the same run. The calculation is explained in more detail here.
How do I select this option?
To use this method, select Absolute Quantitation for HCP using Hi-N at the Protein options dialog at the Resolve Conflicts or Review Proteins screens:
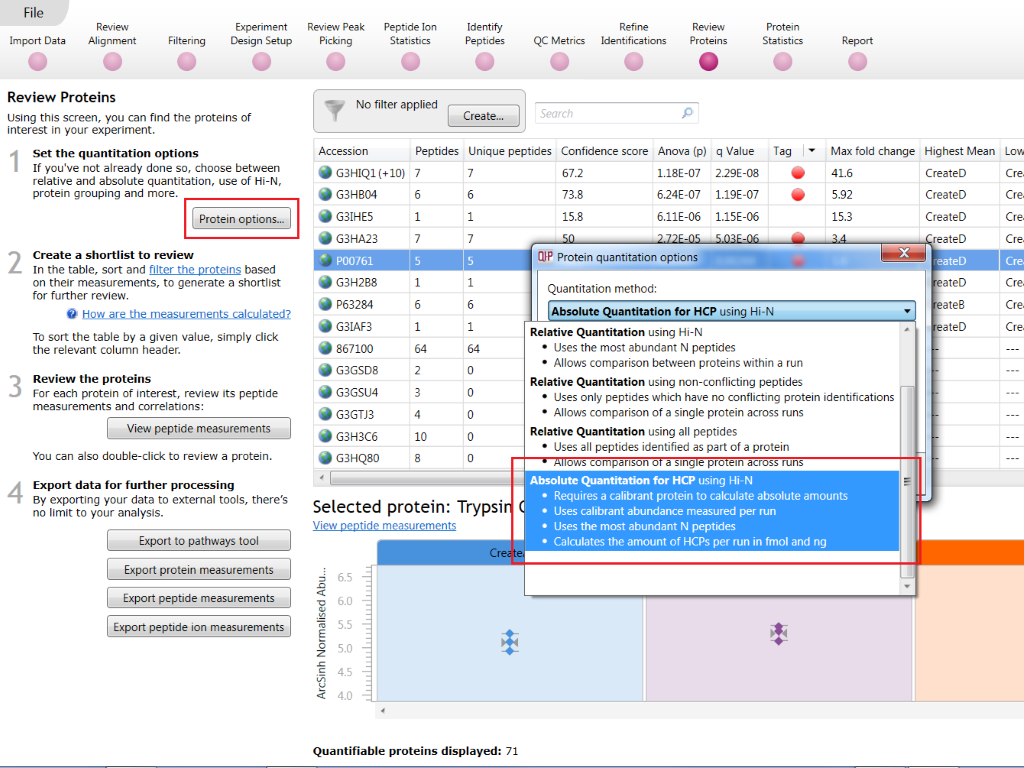
Selecting the Absolute Quantitation for HCP using Hi-N option at Review Proteins
Specify the calibrant protein by accession, the amount present (in fmol), the number of Hi-N peptides to use for the calculation, and whether to carry out protein grouping.
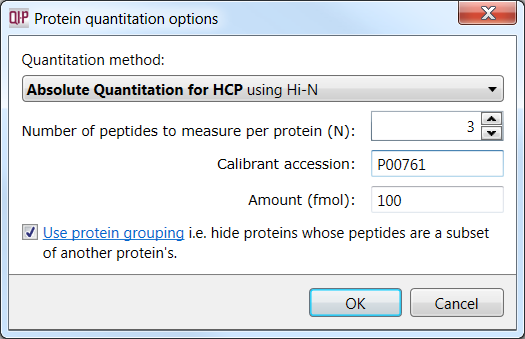
Selecting the calibrant
The software will then calculate the amounts (fmol and mass in ng) of all identified proteins present. In the table at Review Proteins, these are presented as averages by group:
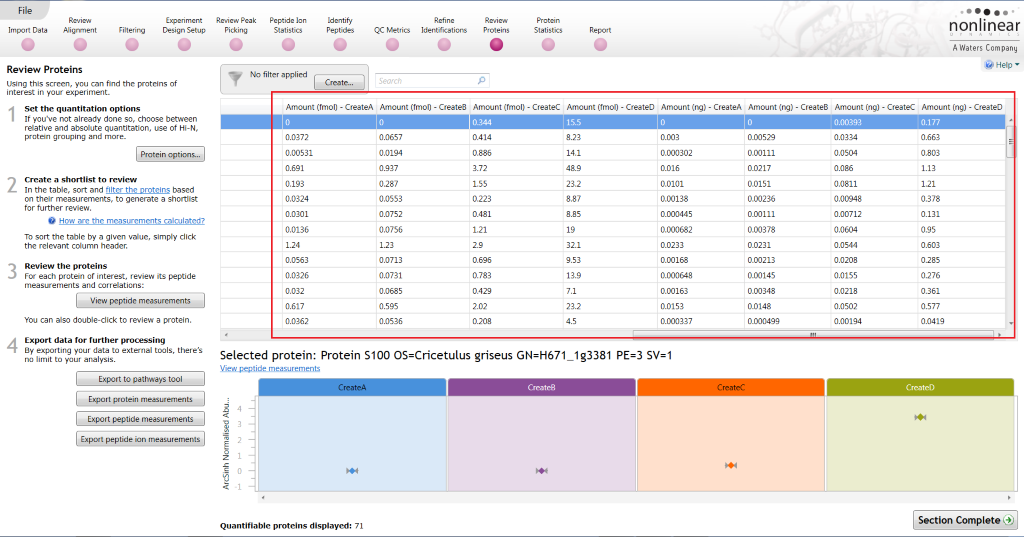
Where to view the calculated values
You can also select these for exporting via Export protein measurements:
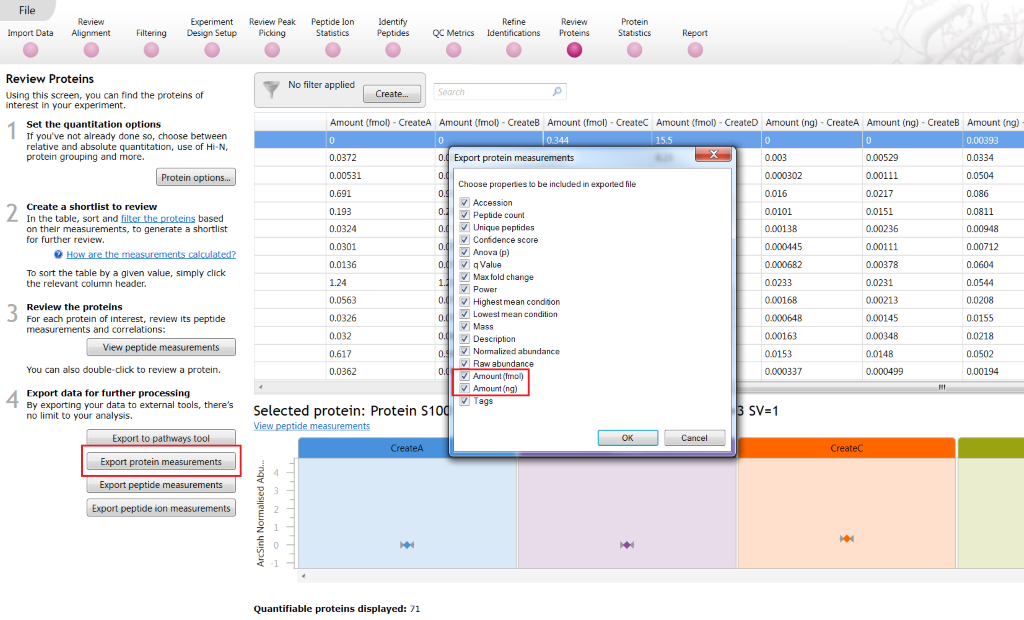
Exporting run-by-run values
In the .csv export resulting from this, the fmol and ng values are provided on a run-by-run basis alongside the usual measurements.
Please note that for the ng mass of protein to be calculated, you must have identified the protein from a database entry with a value for the protein molar mass. If this is not present, you will only see the fmol results for the calculation for that protein. Equally, if the mass in the database is altered or incorrect, that will affect the value calculated for that protein.
How do I select this option in a fractionated experiment?
If you are analysing fractionated samples, you can select this quantitation option at the Review Proteins stage of the workflow as usual, after recombining your samples. The calculation for multi-fraction experiments is slightly different to that for unfractionated experiments, and is explained here.
Should I use the therapeutic protein as the calibrant?
Whilst you can do this, it is likely to be on a very different scale to the contaminants, quantitatively speaking. We recommend that you spike in a calibrant to each run that is more comparable to the dynamic range of the contaminants that you are intending to quantify.
See also
- How does Hi-N work?
- How is absolute quantitation by Hi-N for HCP analysis calculated?
- How does Hi-N for HCP analysis work in the fractionation workflow?
- When using absolute quantification by Hi-N, why are the calculated amounts of my calibrant protein not equal to the value I enter?
- What happens to protein measurements when the calibrant protein can't be found?