




Which quantitation method should I choose for my experiment?
Progenesis QI for proteomics allows you to select from five approaches to protein-level quantitation. These can be accessed using the Protein options dialog at the Resolve Conflicts or Review Proteins windows. A drop-down is available showing the following options:
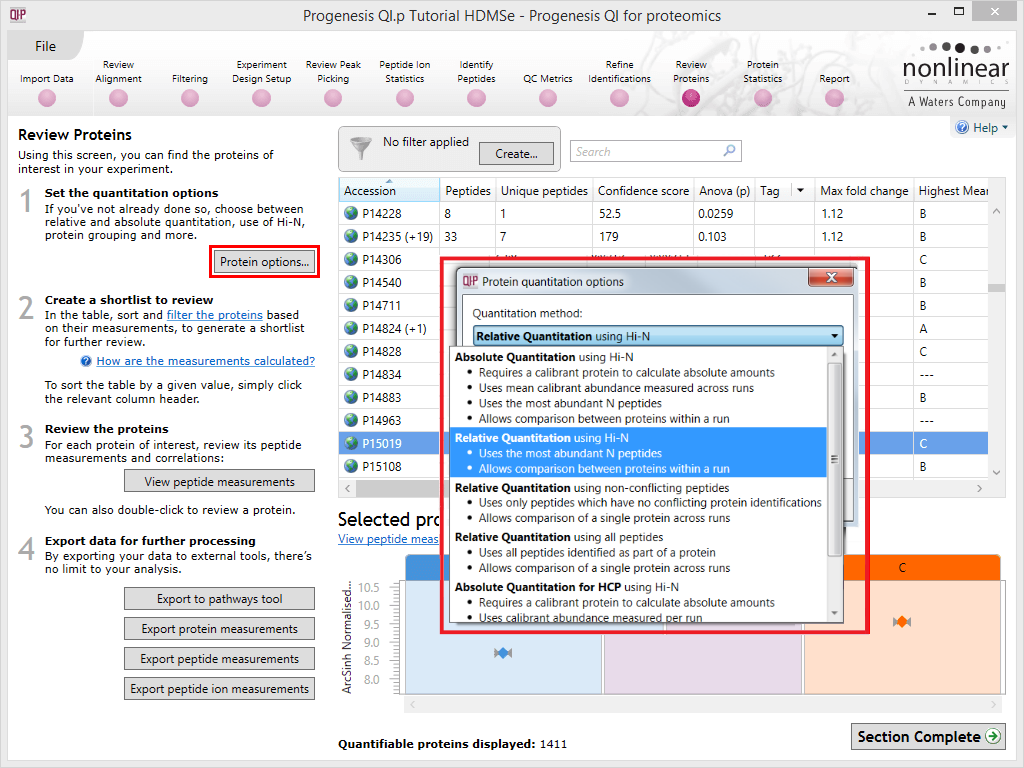
The Protein options dialog with the options drop-down activated.
The approaches are as follows; these descriptions should assist you in selecting an approach.
Absolute quantitation using Hi-N
This option will apply Hi-N using an absolute calibrant, which must have been loaded at the same level in every run and identified. In this case, the N most abundant peptides for each protein have their individual abundances averaged to provide a protein abundance reading. The abundance of the calibrant under this scheme and its known loading amount are used to generate a relationship between amount and abundance. This is then applied to all proteins, determining absolute amounts of every protein in every run, and allowing direct comparisons between the same protein in different runs and different proteins within a run. More information on Hi-N is available in a dedicated FAQ page.
Relative quantitation using Hi-N
This option will apply Hi-N without an absolute calibrant. In this case, the N most abundant peptides for each protein have their individual abundances averaged to provide the protein abundance reading. This value is then compared relatively across runs; since the same peptides are being compared in each case, this is a fair comparison, with no need for a calibration to allow protein-to-protein comparison within a run. More information on Hi-N is available in a dedicated FAQ page.
Relative quantitation using non-conflicting peptides
Proteins are quantified using only peptides that are not also part of another protein hit; that is, protein abundance in a run is calculated from the sum of all the unique normalised peptide ion abundances corresponding to that protein.
The discounting of conflicted peptides may sacrifice data, but using unambiguously assigned peptides provides a more confidently specific read-out of protein abundance, preventing confounding effects such as the overlapping of trends derived from different proteins.
The process is illustrated below.
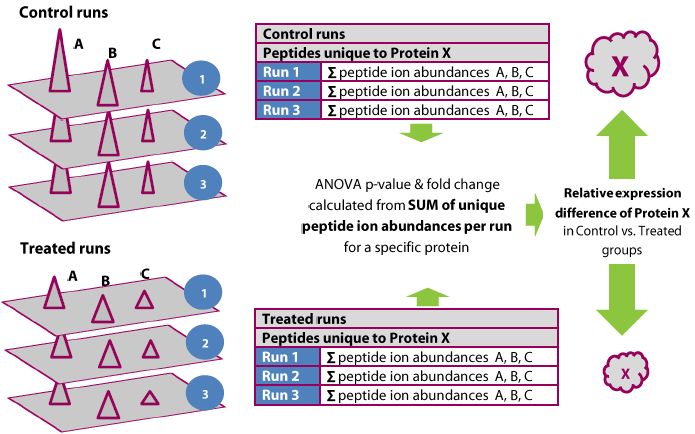
The quantitation is relative and over the same species in every case (aided by the accurate alignment and co-detection employed by Progenesis). In effect, the assumption is made that if protein X is in the ratio A:B between two samples, then any of its constituent peptides are also in that ratio A:B, and so not all peptides are essential to determine this ratio – and more specifically, that only peptides unambiguously reflecting that protein's ratio should be used. However, the more peptides that are measured, the more accurate the reading.
The use of the summed abundance also increases the weighted effect of more abundant peptide ions, with a better signal to noise ratio.
Peptide conflicts can be resolved at the Resolve Conflicts stage of the workflow if desired (see the user guide) to assign ambiguous peptides to one protein hit or another. Also, the protein grouping FAQ describes an important factor regarding the unique peptides only option; homologous identities or multiple database entries for one protein could mean a large number of 'spurious' conflicts appear, that would affect the peptides available for quantitation. Enabling protein grouping addresses this issue.
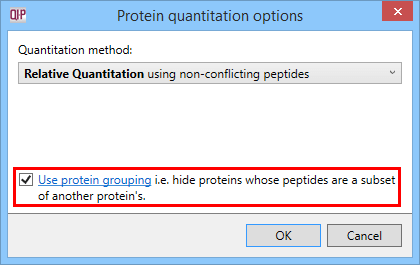
The Protein options dialog with the protein grouping selection highlighted.
Finally, it should be noted that using only unique / non-conflicting peptides may remove some proteins entirely from quantitation, if all their peptides have conflicts. This may affect the number of proteins reported by the software as being quantifiable at the base of the Review Proteins window.
Relative quantitation using all peptides
Proteins are quantified using all peptides. This does not sacrifice any data, but is more at risk of confounding effects from the overlap of trends derived from different proteins. If all conflicts had been resolved, then this approach would not suffer from such a problem. In practice, this may not be case, and this option should be employed with caution. Also, the number of quantifiable proteins may be altered; in this method, all possible proteins will appear, while some may be eliminated from the non-conflicting-only method.
Absolute quantitation for HCP using Hi-N
This option will also apply Hi-N with an absolute calibrant, but with some modifications made to suit Host Cell Protein (HCP) analyses. More information on Hi-N for HCP is available in a dedicated FAQ page.
See also
- How does Hi-N work?
- How does Hi-N work in the fractionation workflow?
- When using absolute quantification by Hi-N, why are the calculated amounts of my calibrant protein not equal to the value I enter?
- How does absolute quantitation by Hi-N for HCP analysis work?
- How does Hi-N for HCP analysis work in the fractionation workflow?