





How does Progenesis correct for drifts in retention time?
In Progenesis QI, one run is chosen as the alignment reference and all other runs have their retention times aligned to the reference. This allows us to correct for any drift in retention time. This gives adjusted retention times that are equivalent across all runs. Then, during peak picking, we can create a single set of compound ion outlines, shared by all runs.
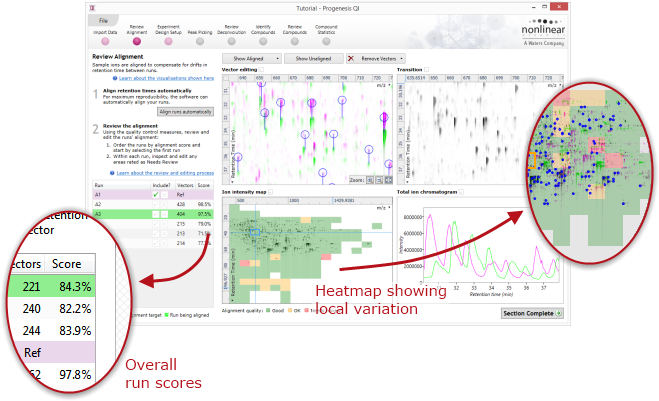
Alignment is driven by the placement of landmarks called alignment vectors. Each vector connects the location of a particular compound ion on the reference run with the location of the same ion on the run being aligned. Alignment can be performed automatically, or vectors can be placed manually. Once the vectors are placed they are used to calculate a non-linear mapping between the retention times of the reference run and those of the run being aligned.
Automatic alignment
When using automatic alignment, each run in the experiment is aligned to experiment's alignment reference run separately. An initial gross alignment is calculated by finding a small number of retention time shifts which bring most of the chromatogram into close alignment.
The result of this initial alignment is then fine-tuned by making a series of small, tentative adjustments and assessing their impact on the overall quality of alignment. The measures of the quality of the final result can be seen at Review Alignment, both in the list of runs and in the alignment quality heatmap (show me).
{kind=link}
If any alignment vectors have already been added manually before the automatic alignment process is started, these vectors are used as a starting point for the initial gross alignment.